Lessons 38 and 39: Quantifiying gene expression¶
The goal of this unit is to learn how to compute simple quantitiative metrics from next-generation sequencing data. In particular, we will use RNA-Seq data and quantify gene expression using a package called Pysam, a python package that wraps the popular Samtools package.
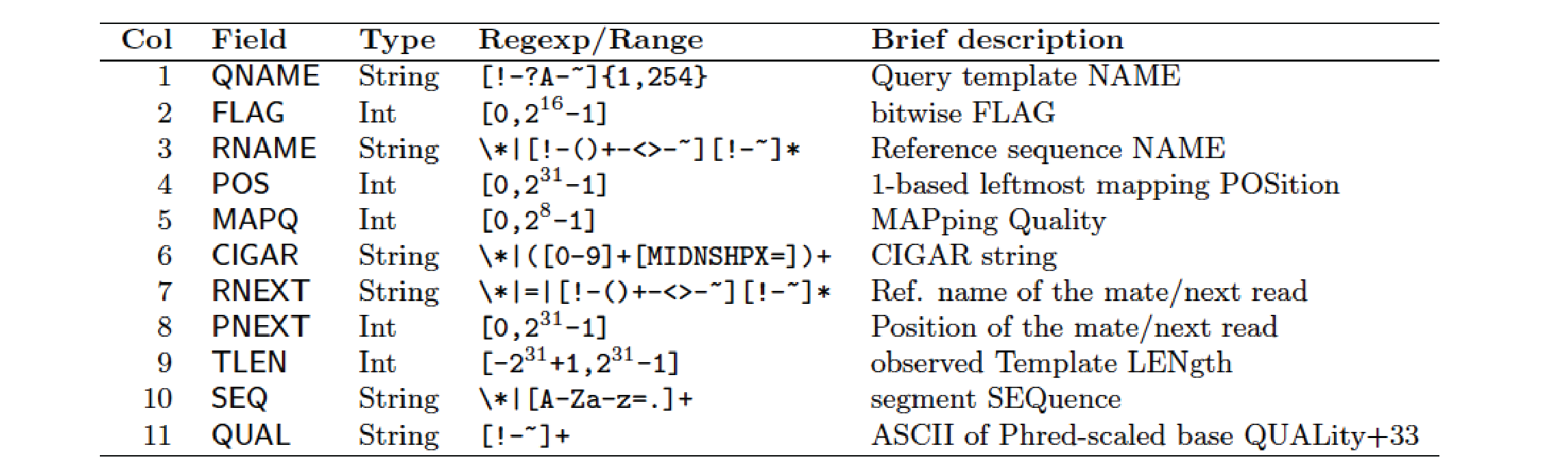
A file format called SAM/BAM (described here) is now the standard formats for next-generation sequence alignments.
The Samtools API (link) provides a good description of some of these methods. Pysam is a Python package that wraps these tools and enables many useful manipulations of SAM/BAM files.
First, we will need to install Pysam, which can be done by:
pip install pysamBefore we start working with Pysam, let's start with some simple methods to compare two features. We say that 2 intervals overlap if any region of the two features cover the same position.
def overlap(x, y):
"""
This function takes two intervals and determines whether
they have any overlapping segments.
"""
new_start = max(x[1], y[1])
new_end = min(x[2], y[2])
if new_start < new_end and x[0] == y[0]:
return True
return False
region1 = ("chr3", 27, 82)
region2 = ("chr4", 27, 82)
region3 = ("chr3", 57, 75)
region4 = ("chr3", 85, 107)
Region 1 does NOT overlap region 2 because it is on a different chromosome.
overlap(region1, region2)
But, it does overlap region 3.
overlap(region1, region3)
But, not region 4 which is after it.
overlap(region1, region4)
# Sox2 gene location in mouse genome
gene=("chr3", 34548926, 34551382)
Once Pysam is installed, you can load the package by simply typing:
import pysam
To start exploring reads from a SAM/BAM file, let's first load the file.
# load BAM file
bamfile = pysam.AlignmentFile('DGE5p.bam', 'rb')
Note the 'rb' parameter indicates to read the file as binary, which is the BAM format (BAM stands for Binary Alignment Map). If loading a SAM file, this parameter does not need to be specified
An important and very common application is to count the number of reads (aligned fragments) that overlap a give feature (i.e. region of the genome or gene). One simple approach to doing this is to make a list of all reads generated and simply iterate over the reads to identify whether a read overlaps a region. In this case we need an overlap method that can compare a simple interval (defined by a tuple of chr, start, end) with a pysam AlignedSegment object. This overlap method needs the AlignmentFile object to decode chromosome name, otherwise it is very similar to the above overlap() function.
def overlap(x, gene, bamfile):
"""
A modified version of overlap that takes an interval and a pysam
AlignedSegment and tests for overlap
"""
new_start = max(x.reference_start, gene[1])
new_end = min(x.reference_end, gene[2])
if (new_start < new_end and bamfile.getrname(x.tid) == gene[0]):
return True
return False
Let's get all of the reads in the BAM file
bam_iter = bamfile.fetch()
Now, let's iterate through all of these reads and count whenever a read overlaps our gene of interest
# code to iterate over reads and count for a single gene
naive_gene_count = 0
for x in bam_iter:
# Note x is of type pysam.AlignedSegment
if(overlap(x, gene, bamfile)):
naive_gene_count += 1
Be careful, this takes a long time to run...
Let's now print this count
print(naive_gene_count)
The problem with this approach is that to do this, you will need to store the entire file in memory. Worse than that, in order to find the reads within a single gene, you would need to iterate over the entire file, which can contain hundreds of millions of reads. This is quite slow even for a single gene, but will only increase if you want to look at many genes.
Instead, more efficient datastructures (and indexing schemes) can be used to retrieve reads based on positions in more efficient ways.
Now that you have access to all of the methods in the Pysam package. Let's take a tour of some of the things you can do in Pysam. We'll start by counting the number of reads over the same region above, but in a smarter way.
(Note: the exact way Pysam, Picard and other tools implement this is based on a similar concept to the interval tree, but generally involve a tree built on a file index so as to avoid reading the entire file into memory, but for the purposes of this discussion the implementation details are not important).
To get the reads overlapping a given region, Pysam has a method called fetch(), which takes the genomic coordinates and returns an iterator of all reads overlapping that region.
bam_iter = bamfile.fetch(gene[0], gene[1], gene[2])
You can now access each alignment contained in the iterator and perform any functions you want on these. For now, we will simply count them
pysam_gene_count = 0
for x in bam_iter:
pysam_gene_count += 1
This is the same thing we did earlier and we can compare the results
print(pysam_gene_count)
Not surprisingly, they are the same - the big difference is that the second method is significantly faster. The reason for this is that Pysam (and Samtools, Picard, and other similar packages) make use of clever tricks to index the file by genomic positions to more efficiently search for reads within a given genomic interval.
Now that we know how to count reads overlapping a region, we can write this as a function and try and compute this for all genes.
def read_count(gene, bamfile):
"""
Compute the number of reads contained in a bamfile that overlap
a given interval
"""
bam_iter = bamfile.fetch(gene[0], gene[1], gene[2])
pysam_gene_count = 0
for x in bam_iter:
pysam_gene_count += 1
return pysam_gene_count
You can run this with any gene tuple now
gene2=("chr19", 584229, 5845480)
read_count(gene2, bamfile)
Now, let's compute the gene counts for all genes. We'll start by reading in all genes
gene_counts={}
with open('chr3.genes.bed', 'r') as f:
for line in f:
tokens = line.split('\t')
gene_local = (tokens[0], int(tokens[1]), int(tokens[2]))
count = read_count(gene_local, bamfile)
gene_counts.update({tokens[3] : count})
Now let's query the gene counts for Sox2 (ID: NM_011443)
print(gene_counts.get("NM_011443"))
print(gene_counts.get("NM_016904"))
print(gene_counts.get("NM_001038696"))
In practice, the number of reads overlapping a gene is never used to quantify gene expression (because read numbers are dependent on gene expression as well as gene length). A more common metric for gene expression is called the RPKM. RPKM is a fairly simple transformation of read counts that normalizes the number of counts by gene length as well as the average number of reads per base in the genome.
gene_length = gene[2] - gene[1]
print(gene_length)
total_mapped_reads = 0
# here you want all reads
bam_iter = bamfile.fetch()
for x in bam_iter:
total_mapped_reads += 1
print(total_mapped_reads)
rpkm_metric = 1e9 * read_count(gene, bamfile) / (total_mapped_reads * gene_length)
print(rpkm_metric)
We can now rewrite the quantification method to compute RPKMs for all genes
def rpkm(genefile, bamfile):
"""
Compute the RPKM (reads per kilobase per million mapped reads) metric for all genes within the genefile using
an RNA-Seq bamfile
"""
total_mapped_reads = 0
# here you want all reads
bam_iter = bamfile.fetch()
for x in bam_iter:
total_mapped_reads += 1
geneCounts={}
with open(genefile, 'r') as f:
for line in f:
tokens = line.split('\t')
gene_local = (tokens[0], int(tokens[1]), int(tokens[2]))
gene_length=gene_local[2]-gene_local[1]
rpk_metric = 1e9 * read_count(gene_local, bamfile) \
/ (total_mapped_reads * gene_length)
gene_counts.update({tokens[3] : rpk_metric})
return(gene_counts)
Now, lets give it a try
gene_counts = rpkm('chr3.genes.bed', bamfile)
print(gene_counts.get('NM_011443'))